Using GRiTS on simulation data¶
In the following example we will demonstrate using GRiTS on data from a real simulation. It is advisable to go through the “Intro to GRiTS” tutorial first.
import numpy as np
import mbuild as mb
from grits import CG_System
from grits.utils import amber_dict
Coarse-graining¶
The following example will use a GSD file from a HOOMD simulation initialized and run using PlanckTon.
The simulation was run using the general amber forcefield and the chemistries are shown below:
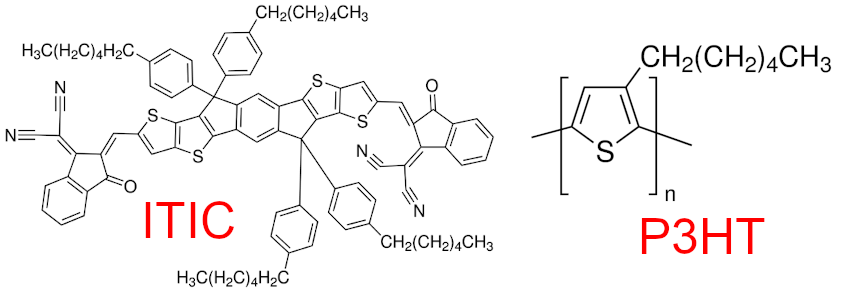
structures¶
In order to apply a coarse-grain mapping to an atomistic structure we need to know the atom indices which correspond to each bead. GRiTS works by using SMILES strings to find instances of that chemical fragment and map those atom indices to a bead. The goal of this example is to map the backbone of each ITIC molecule and the thiophene ring in each P3HT monomer (the GSD file contains a 16-mer) to a bead.
The next cell visualizes the target fragments:
itic_backbone = "c1c4c(cc2c1Cc3c2scc3)Cc5c4scc5"
thiophene = "c1cscc1"
a = mb.load(itic_backbone, smiles=True)
a.visualize().show()
b = mb.load(thiophene, smiles=True)
b.visualize().show()
Next we will pass these SMILES patterns, along with our gsdfile and type
dictionary, to the CG_System class to create our coarse-grained
structure. The SMILES patterns for these beads are chosen carefully such
that they are exclusive: The thiophene moiety used to map P3HT also
exists in ITIC, but because the itic_backbone pattern we have chosen
uses up the atoms in the thiophenes and this pattern is first in the
beads dictionary, only the itic_backbone pattern will match in
ITIC.
The SMILES matching used in the CG_Compound class is slow, so the
CG_System class relies on a couple assumptions:
Molecules (freud.cluster.Clusters determined using bonds) having the same number of particles are assumed to have the same structure. (e.g., if two molecules each have 402 particles, they are assumed to have the same structure.)
Atom order within each structure is assumed to be constant. (e.g., if the order of atoms in water is HOH, then all water structures are in that order–not OHH or HHO.)
The particle indices are assumed to be constant for each frame of the trajectory.
gsdfile = "../grits/tests/assets/itic-p3ht.gsd"
system = CG_System(
gsdfile,
beads={"_A": itic_backbone, "_B" : thiophene},
conversion_dict=amber_dict
)
The cell above will have a couple warnings which can be safely ignored:
one from OpenBabel, another telling us that some atoms were left out of
coarse-graining (expected since we do not map the entire structure of
ITIC or P3HT), and another telling us that the itic_backbone string
wasn’t found (we do not expect this pattern to exist in P3HT).
Next we can save the mapped structure to a GSD file; by default every frame will mapped and saved:
cg_gsd = "itic-p3ht-cg.gsd"
system.save(cg_gsd)
To see the coarse-grain mapping we can open both files in OVITO or VMD with the gsd-vmd plugin.
The images below showing the bead overlaid with the atomistic structure were created using OVITO:
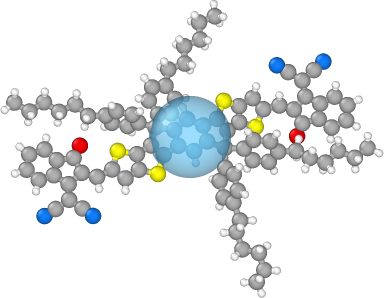
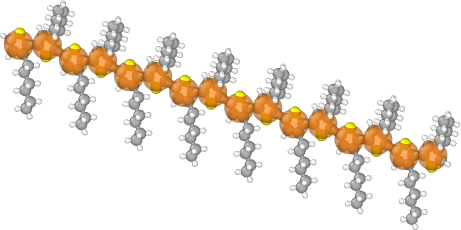
If we have multiple statepoints with the same initial structure and want to reuse our mapping, we can save the mapping to a json file:
cg_json = "itic-p3ht-cg.json"
system.save_mapping(cg_json)